Advancements in the field of molecular dynamics simulations have always been hindered by the complex interactions between atoms and electrons within molecules. However, researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google DeepMind have recently made a groundbreaking discovery in machine learning algorithms that could revolutionize the way we simulate molecules.
The traditional methods of computing electron interactions rely on solving the Schrödinger equation, which can be extremely time-consuming, especially for molecules containing more than a few dozen atoms. This limitation has made it difficult to run molecular dynamics simulations over long time-scales, as the computational cost quickly escalates beyond available resources.
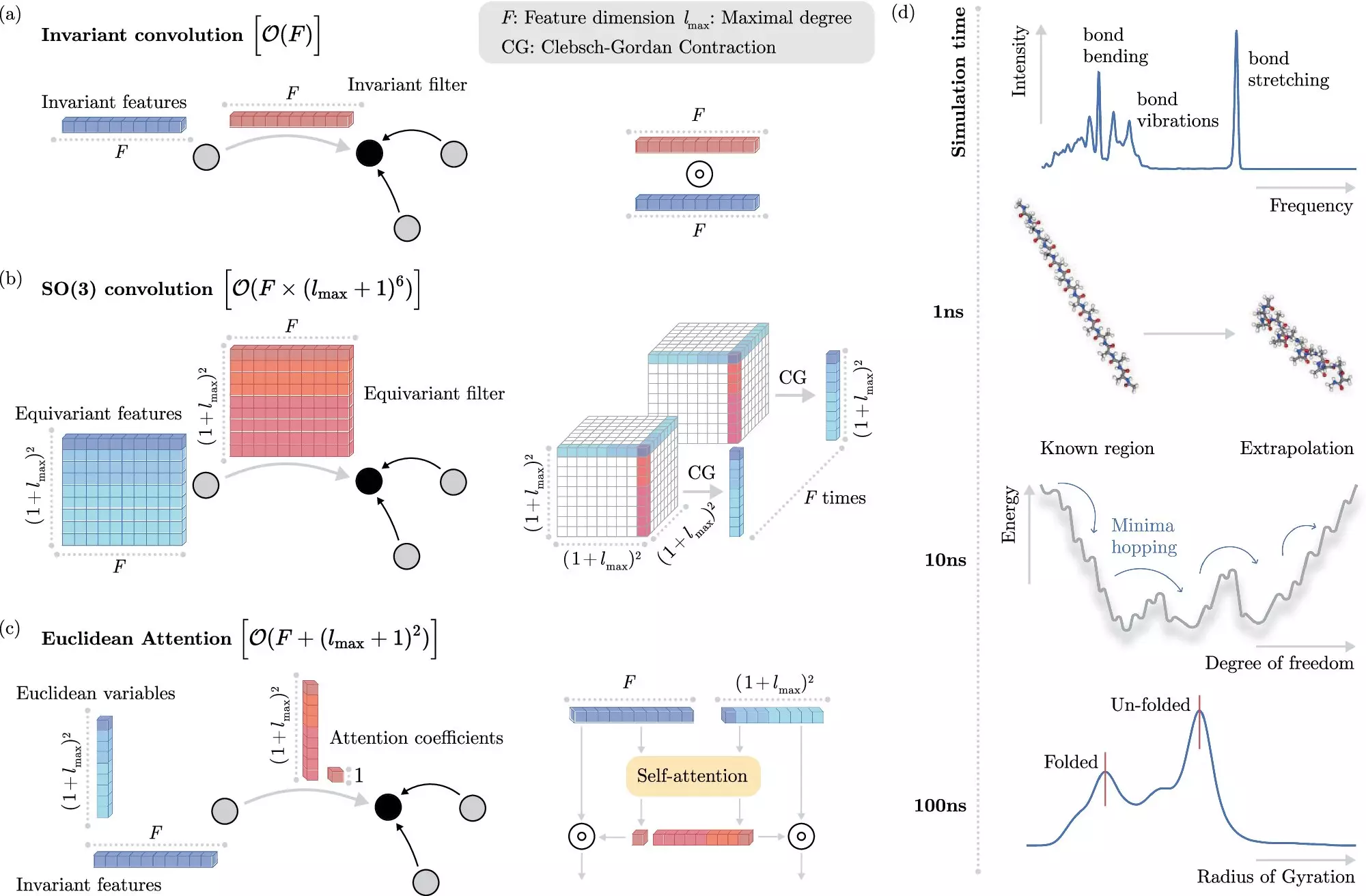
In recent years, machine learning methods have emerged as a potential solution to this problem. Rather than solving the Schrödinger equation explicitly, these algorithms can predict electronic interactions at the atomistic level with reduced computational cost. The challenge, however, lies in teaching the machine learning system to understand these interactions without explicit modeling of the electrons.
The researchers at BIFOLD have developed a new learning algorithm that decouples invariances from other information about a chemical system, simplifying the process and reducing computational cost. This approach allows the machine learning model to focus on the most complex operations that truly matter, improving efficiency and enabling long-time scale simulations.
The ability to accurately simulate the interaction of molecules with proteins in the human body could revolutionize drug development, eliminating the need for expensive and time-consuming experiments. This breakthrough could also have significant implications for material design, such as the development of more efficient solar panels and batteries.
To demonstrate the potential applications of the new machine learning method, the research team used it to identify the most stable version of docosahexaenoic acid, a primary structural component in the human brain. This task would have been infeasible using traditional quantum mechanical methods, highlighting the efficiency and accuracy of the new algorithm.
The success of this research opens up new possibilities for combining advanced machine learning techniques with physical principles to overcome longstanding challenges in computational chemistry. The next generation of algorithms will need to accurately simulate larger and more complex systems, including additional long-range physical interactions.
The breakthrough in machine learning algorithms for molecular dynamics simulations represents a significant advancement in the field of computational chemistry. By combining the power of artificial intelligence with the principles of quantum mechanics, researchers are now able to perform simulations that were previously thought to be impossible. This innovation has the potential to transform the way we understand and manipulate atoms and molecules, leading to new discoveries in drug development, material design, and beyond.
Leave a Reply